

HEMOGLOBIN/ Synthesis/ Degradation/ Forms / Complexes/ Related Disorders.
 |
| HEMOGLOBIN/ Synthesis/ Degradation/ Forms / Complexes/ Related Disorders. |
- HEMOGLOBIN (Hb),
- Synthesis of Hemoglobin
- Degradation of Hemoglobin
- Forms of Hemoglobin
- Complexes of Hemoglobin
Structurally - Abnormal Hemoglobin and Related Disorders
HEMOGLOBIN (Hb)
Introduction:- Structure There are about 2-3 million Hb molecules in a red cell, each having a molecular weight of 64,458.
Introduction:- Structure There are about 2-3 million Hb molecules in a red cell, each having a molecular weight of 64,458.
Hemoglobin is a chromoprotein consisting of a globin molecule attached to 4 red-colored haem molecules. The globin molecule consists of two alpha and two beta polypeptide chains. Haem is a metal complex containing an iron atom in the center of a porphyrin structure.
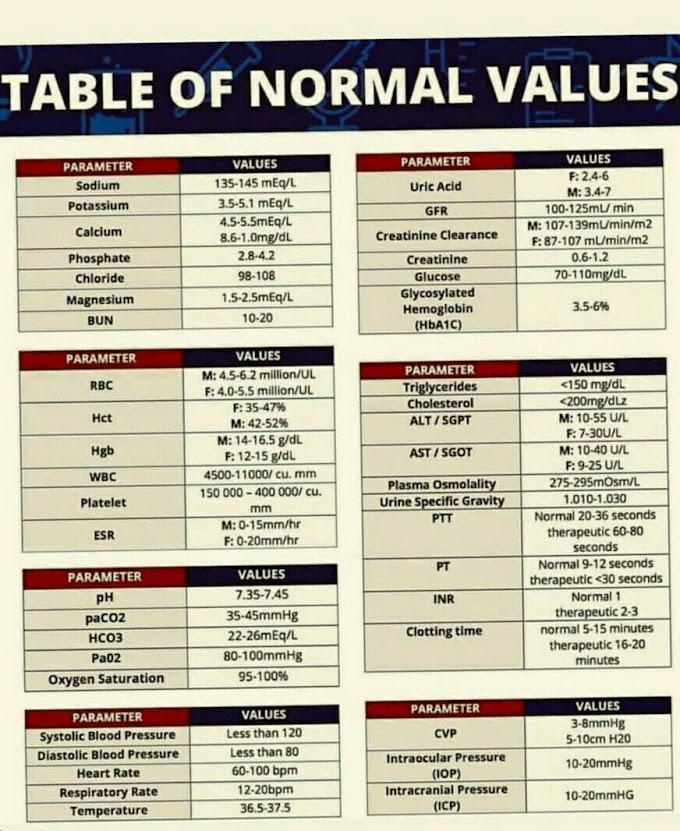
Hemoglobin is formed in the developing erythrocyte in the bone marrow. The life span of hemoglobin is the same as the life span of the red cell that accommodates it, that is, about 120 days. In the adult, the concentration of hemoglobin in the blood is 14-16 grams per 100 ml. A healthy adult loses and produces 6.25 g of hemoglobin per day. A liter of normal blood is capable of carrying 200 ml of oxygen and 1.0 grams of hemoglobin combines with 1.34 ml of oxygen. Oxidization converts haem into haematin. The iron content of each hemoglobin molecule is 3.4 nanograms.
 |
| structure of hemoglobin |
Synthesis of Hemoglobin
Haem synthesis occurs largely in the mitochondria by a
series of biochemical reactions. Glycine + succinyl coenzyme
- A is condensed by delta amino laevulinic acid (ALA) synthetase in the presence of pyridoxal phosphate (Vit B).
- This leads to the formation of protoporphyrin.
Reaction
Glycine +
Vit. B6 + Succinyl CoACALA synthetase Protoporphyrin
2. Protoporphyrin combines with iron to form haem. Fe +
Protoporphyrin— Haem
3. Each molecule of haem combines with the globin chains on polyribosomes.
3. Each molecule of haem combines with the globin chains on polyribosomes.
4. Tetramer of 4 globin chains, each with its own haem
group.
Degradation of Hemoglobin
When the red cell completes its life span, at the end of
about 120 days, it is removed by the macrophages of the reticuloendothelial
system. The hemoglobin is released, broken down, the useful portion is
recirculated and the rest is excreted.
The degradation of hemoglobin begins in the
reticuloendothelial cell. The protein fraction is separated and broken down into
amino acids that enter the common metabolic pool. Iron is separated and stored
as ferritin and haemosiderin for reutilisation in the synthesis of hemoglobin
and other haem proteins. The porphyrin portion is converted into biliverdin in
reticuloendothelial cells and enters the blood as free bilirubin.
In the liver it is conjugated and excreted in bile. In the intestine, bilirubin is converted into faecal urobilinogen by the action of intestinal bacteria. Some faecac urobilinogen is reabsorbed and carried back to the liver to be excreted into the bile. Some escape the liver and is excreted in the urine.
In the liver it is conjugated and excreted in bile. In the intestine, bilirubin is converted into faecal urobilinogen by the action of intestinal bacteria. Some faecac urobilinogen is reabsorbed and carried back to the liver to be excreted into the bile. Some escape the liver and is excreted in the urine.
Forms of Hemoglobin
Depending on the content and sequence of amino acids in the
globin chains, different forms of hemoglobin may occur in the red cells.
Hemoglobin A (HbA)
This is the main adult hemoglobin (97 %) and contains 2 alpha and 2 beta goblin chains. Another form of adult hemoglobin, HbA,, contains 2 alpha chains paired with 2 delta chains. HbA2 is present in normal adults in small amounts (2-4%).Hemoglobin F (HbF)
HbF is the major form of hemoglobin during intrauterine life and at birth, In HbF, 2 alpha chains are paired with 2 gamma chains. HbF is slowly replaced by HbA and at the end of the first year, the HbF concentration is reduced to 1-2 % from 70-90 % at birth. A normal adult may have 1-2% HbF in blood.Hbf combines more readily with oxygen than HbA, thus helping the fetus in acquiring oxygen in placental circulation. It also differs from HbA in that, it resists denaturation with alkali, unlike HbA which is easily denatured.
Complexes of Hemoglobin
Certain complexes of hemoglobin may be formed if it is
acted upon by other chemicals. Such complexes may drastically reduce its
capacity to react with oxygen, thus leading to hypoxia or cyanosis. Such forms
include:
Carboxyhaemoglobin (HbCO)
This is produced when hemoglobin combines with carbon monoxide. Hemoglobin has a much greater affinity for carbon monoxide than for oxygen and combines readily with it. It is found in higher concentrations in the blood of smokers.Methaemoglobin (HI)
The formation of methemoglobin results from its reaction with certain drugs such as phenacetin or sulphonamides. In this form, iron is oxidized from the ferrous to the ferric state and is therefore incapable of reacting with oxygen.Sulphhaemoglobin (SHD)
SHB is formed irreversibly by the action of certain drugs and chemicals such as sulphonamides. Once formed, it remains for the life of the carrier RBC and is not capable of oxygen transport.Structurally Abnormal Hemoglobins and Related Disorders
The term abnormal hemoglobin is reserved for abnormalities
in the globin molecules of hemoglobin. These consist of the formation of abnormal
alpha or beta-peptide chains because one or more of the constituent amino acids
has been substituted by another. Abnormal hemoglobins formed in this way are
designated as hemoglobin C, D, E, G, H, I, J, K and S. These can be
differentiated by techniques of electrophoresis and chromatography.
Abnormal hemoglobins are associated with congenital and
hereditary defects (inborn errors). The disorders produced by abnormal hemoglobin are called hemoglobinopathies. For instance, possession of hemoglobin S (HS) makes the red cell sickle-shaped in reduced oxygen tension.
In thalassemia, the abnormal gene interferes with the formation of adult hemoglobin (HbA).



If you have any queries related medical laboratory science & you are looking for any topic which you have have not found here.. you can comment below... and feedback us if you like over work & Theory
.
Thanks for coming here..